
While we try to keep things accurate, this content is part of an ongoing experiment and may not always be reliable.
Please double-check important details — we’re not responsible for how the information is used.
Genes
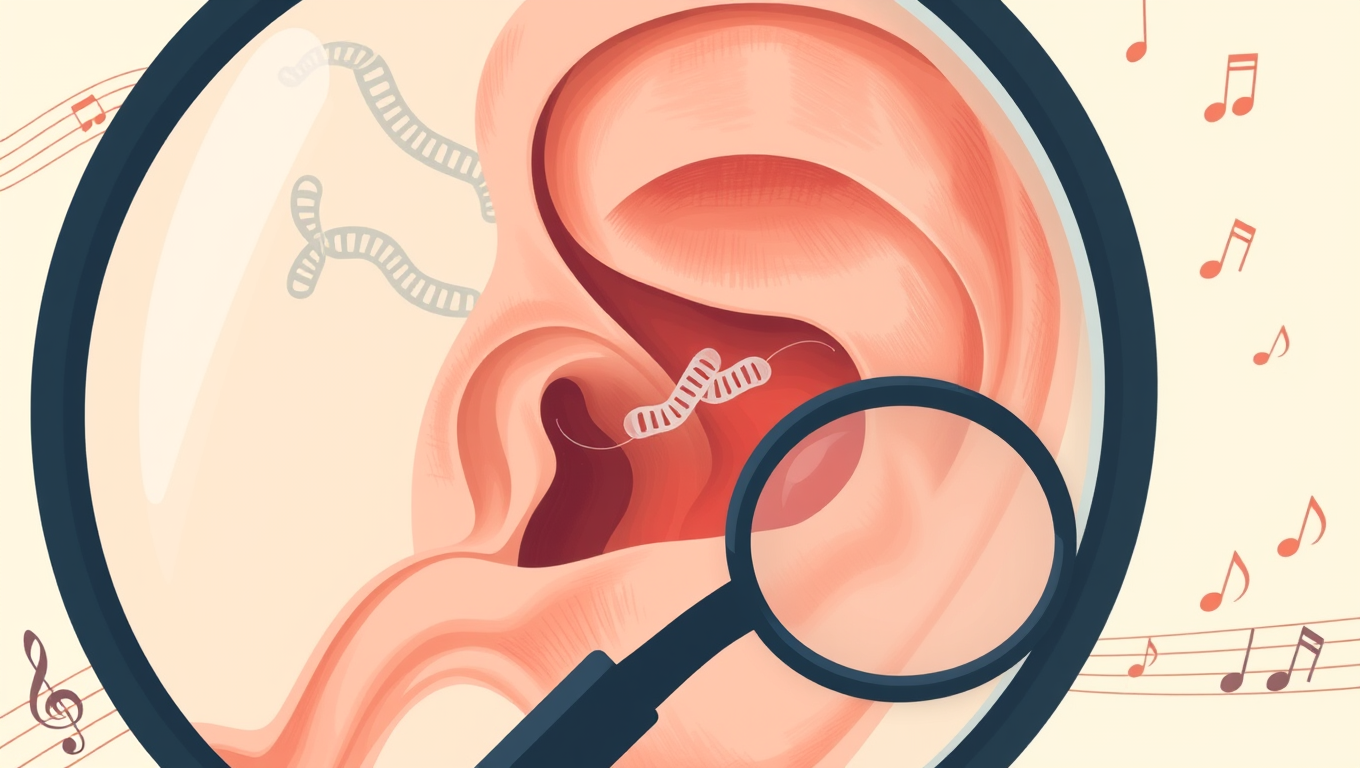
Unveiling the Secrets of Congenital Deafness: New Candidate Genes Revealed
New candidate genes which could be responsible for deafness have been identified.








Artificial Intelligence
Accelerating Evolution: The Power of T7-ORACLE in Protein Engineering
Researchers at Scripps have created T7-ORACLE, a powerful new tool that speeds up evolution, allowing scientists to design and improve proteins thousands of times faster than nature. Using engineered bacteria and a modified viral replication system, this method can create new protein versions in days instead of months. In tests, it quickly produced enzymes that could survive extreme doses of antibiotics, showing how it could help develop better medicines, cancer treatments, and other breakthroughs far more quickly than ever before.

Cold and Flu
“Unlocking the Mystery: Scientists Discover How to Break Down Brain Cell Clumps and Develop New Treatment”
Scientists have discovered how harmful clumps inside brain cells—linked to diseases like ALS and Huntington’s—form, and found a way to break them apart. These sticky tangles of RNA develop inside tiny liquid-like droplets in cells and can linger long after their surroundings vanish. By introducing a special protein, the team could stop the clumps from forming, and with a custom-designed piece of RNA, they could even dissolve them.
Diet and Weight Loss
A New Genetic Test to Predict Obesity Before Kindergarten: A Breakthrough in Preventing Childhood Obesity
A groundbreaking study involving genetic data from over five million people has uncovered how our DNA can predict obesity risk as early as childhood. The new polygenic risk score outperforms previous methods, helping to identify high-risk children before weight issues develop paving the way for early lifestyle interventions.


A New Horizon for Vision: How Gold Nanoparticles May Restore People’s Sight

Retiring Abroad Can Be Lonely Business

Revolutionizing Quantum Communication: Direct Connections Between Multiple Processors

Household Electricity Three Times More Expensive Than Upcoming ‘Eco-Friendly’ Aviation E-Fuels, Study Reveals

“Unveiling Hidden Patterns: A New Twist on Interference Phenomena”
Harnessing Water Waves: A Breakthrough in Controlling Floating Objects
Reducing Falls Among Elderly Women with Polypharmacy through Exercise Intervention
-
 Detectors1 year ago
Detectors1 year agoA New Horizon for Vision: How Gold Nanoparticles May Restore People’s Sight
-
Earth & Climate1 year ago
Retiring Abroad Can Be Lonely Business
-
Cancer1 year ago
Revolutionizing Quantum Communication: Direct Connections Between Multiple Processors
-
Earth & Climate1 year ago
Household Electricity Three Times More Expensive Than Upcoming ‘Eco-Friendly’ Aviation E-Fuels, Study Reveals
-
Chemistry1 year ago
“Unveiling Hidden Patterns: A New Twist on Interference Phenomena”
-
Albert Einstein1 year ago
Harnessing Water Waves: A Breakthrough in Controlling Floating Objects
-
Diseases and Conditions1 year ago
Reducing Falls Among Elderly Women with Polypharmacy through Exercise Intervention
-
Acid Rain1 year ago
“Revolutionizing Honey Bee Survival: A New Pollen-Replacing Food Source Brings Hope for the Future”








